End Users
The Falsified Medicines Directive and the Delegated Regulation specify the verification of safety features and the deletion of unique identifiers by the wholesalers and the entities that are authorised or have the right to supply medicines to the population. According to the Regulation and applicable Latvian regulatory acts, the companies that are authorised or have the right to supply medicines in Latvia are pharmacies, hospitals, clinics, outpatient clinics, health centres, dental clinics and practices.
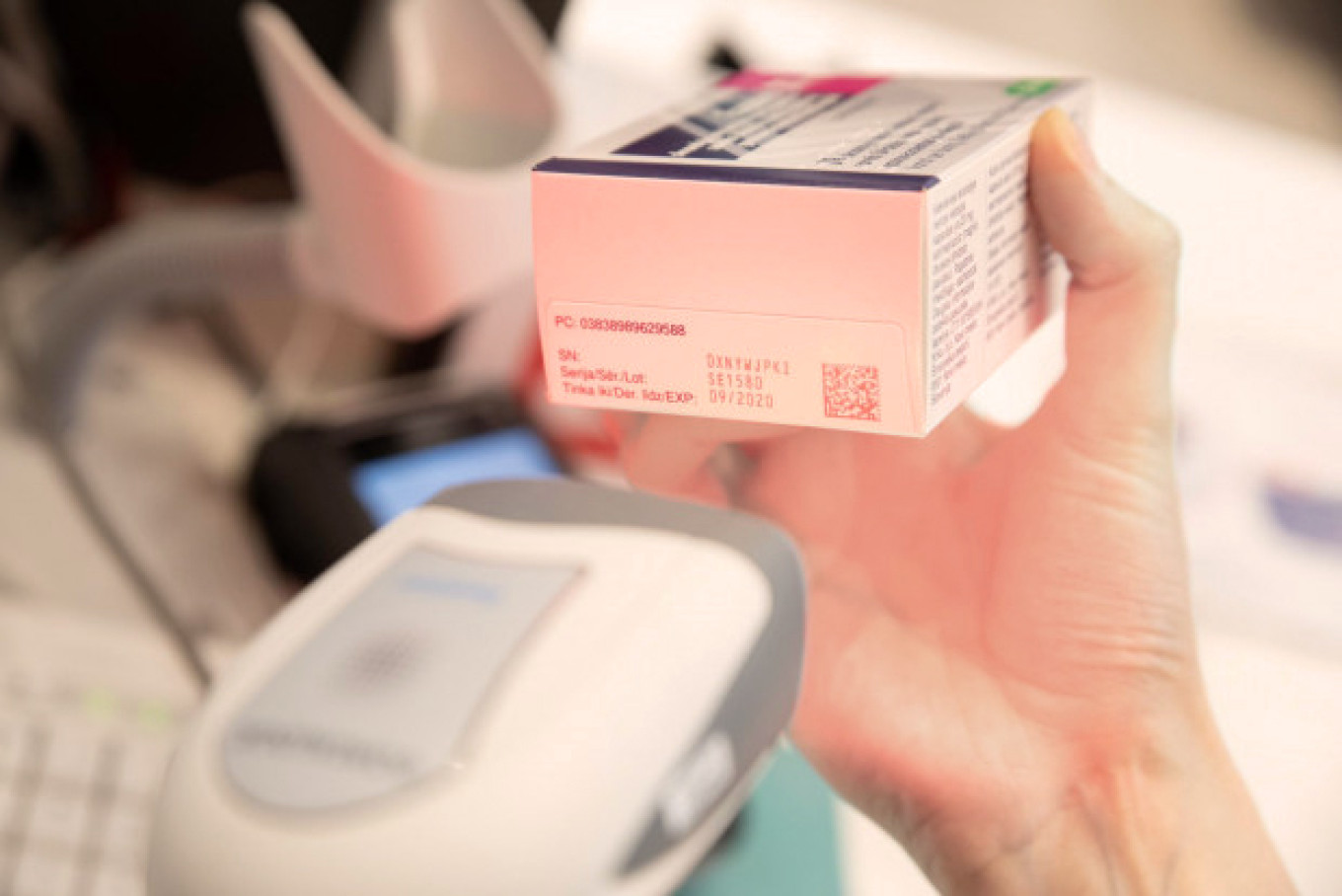
The Latvian Medicines Verification System includes application programming interfaces that wholesalers, pharmacies, hospitals, clinics, outpatient clinics, health centres and medical practices use to send requests to this system, by using their IT software, to verify the authenticity of unique identifiers and to decommission them from the repository system.
The pharmacies and other companies that are authorised and have the right to supply medicines to the Latvian population, are obliged to create an IT connection to the national repository system, as well as to verify the unique identifiers on the packaging and to decommission them from the system with a special technical equipment (a scanner).
From 9th February 2019, pharmaceutical wholesalers across Europe will be required to verify medicines bearing safety features that pass through their warehouses in specific circumstances as set out in the Commission Delegated Regulation (EU) 2016/161. They will also have to decommission packs in certain circumstances, including when they are being supplied to specific customers as per Article 23 of the Delegated Regulation.
A wholesaler must verify the authenticity of medicines on a risk approached basis. This means the wholesaler must verify the unique identifier of at least those products that are returned to him by pharmacies or other wholesalers. Also, products the wholesaler receives from another a wholesaler who is neither the manufacturer nor the wholesaler holding the marketing authorisation nor a wholesaler who is designated by the marketing authorisation holder, must be verified.
Article 20(b) of the Commission Delegated Regulation (EU) 2016/161 relieves wholesalers from the requirement to verify packs if received from the manufacturer or MAH or a wholesaler designated by the MAH, by means of a written contract, to store and distribute products covered by his MA on his behalf (i.e. ‘Designated Wholesaler’). MAHs (including parallel importers / parallel distributors) are obliged to upload to the EU Hub a list of ‘Designated Wholesalers’ for each product placed on the market. For detailed guidance on Designated Wholesalers, please see Appendix 5 of the EMVO Master Data Guide which is available on the EMVO website.
The IT system of each company can be set up by its internal IT employees or outsourced partners. Each company may choose either to integrate the functions of medicines verification and unique identifier deletion into its own IT system or to use them separately. It is a matter to be decided in each company internally.
The LZVO is responsible for developing and supervising the national medicines verification system that receives and stores the information about the safety features of the medicines from the European central database. The LZVO introduces procedures that ensure that only users with a verified identity, role and legitimacy can access the repository or upload information to it.
The competent authorities of Latvia – the State Agency of Medicines and the Health Inspection – supervise the repository to ensure that the requirements of the Delegated Regulation are fulfilled.
DOCUMENTS
The Falsified Medicines Directive is Directive 2011/62/EU passed by the European Parliament and the Council on 8 June 2011 that amends Directive 2001/83/EC on the Community code relating to medicinal products for human use and prevention of falsified medicines from getting into a legal chain of supply
The Delegated Regulation is delegated regulation (EU) 2016/161 of 2 October 2015 supplementing Directive 2001/83/EC of the European Parliament and of the Council by laying down detailed rules for the safety features appearing on the packaging of medicinal products for human use
The Pharmaceutical Law regulates the activity of physical and legal entities in the field of pharmacy, as well as ensures manufacturing and distribution of high-quality and medically suitable medicines of a proper preventive, therapeutical and diagnostic level
Cabinet Regulation No. 220 "Procedures for the Acquisition, Storage, Use, Registration and Disposal of Medicinal Products in Medical Treatment Institutions and Social Care Institutions"
Cabinet Regulation No. 304 "Regulations Regarding the Procedures for the Manufacture and Control of Medicinal Products, the Requirements for the Qualification and Professional Experience of a Qualified Person and the Procedures for the Issuance of the Certificate of Good Manufacturing Practice to a Medicinal Products Manufacturing Undertaking"
Cabinet Regulation No.57 "Regulations Regarding Procedures for the Labelling of Medicinal Products and the Requirements to Be Set for the Package Leaflet of Medicinal Products"
Cabinet Regulation No. 416 "Procedures Regarding the Distribution and Quality Control of Medicinal Products"